LA JOLLA, CA—In addition to the four major bases in the DNA alphabet – A, C, G and T – there is also a minor “fifth” base, 5-methylcytosine (5mC), which plays a disproportionately important role in deciding whether genes and other DNA elements are turned on or off. Not surprisingly, defects in cytosine methylation are associated with developmental abnormalities, genetic diseases and cancer.
In their latest study, published in this week’s online issue of Proceedings of the National Academy of Sciences, researchers at La Jolla Institute for Immunology (LJI), reveal how the finely tuned balance between DNA methylation and demethylation prevents genomic instability and cancer.
While scientists knew that enzymes known as DNMTs were responsible for putting the 5mC mark on cytosines, how this mark was removed remained a mystery for decades. In an earlier study, Anjana Rao, Ph.D., LJI professor and senior author of the current study, together with former colleagues at Harvard University, Cambridge, and the National Institutes of Health, Bethesda, Maryland, showed that cancer-related proteins known as TETs convert 5mC to a “sixth base”, 5hmC, which subsequently reverts to C. Soon thereafter, it became clear that loss of TET function is strongly linked to many types of cancers, both in humans and in mice.
Although DNMTs and TETs are expected to have opposite activities—DNMTs produce 5mC whereas TETs remove it—human blood cancers with mutations in either TET2 or DNMT3A display similar features, including increased levels of DNA damage and genome instability. Moreover, DNMT3A and TET2 are the top two proteins mutated in clonal hematopoiesis, a disease of aging in which certain clones of blood stem cells expand more than others. People with clonal hematopoiesis are at risk for atherosclerosis and for progressive cancers.
The latest findings by the Rao lab provide an explanation why mutations in TETs and DNMTs have similar effects in disease.
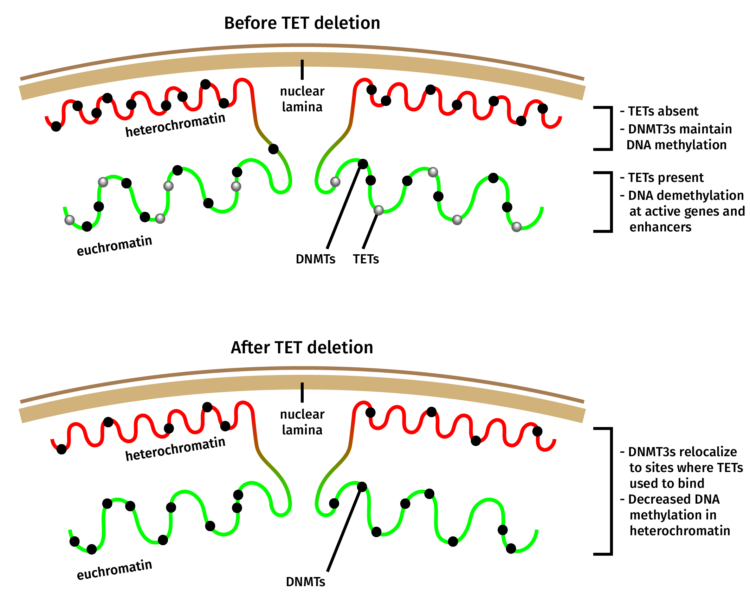
When Isaac F. López-Moyado, a graduate student in the Rao lab and the first author of the study, analyzed the methylation patterns in an aggressive lymphoma that arose when TET2 and TET3 were deleted in mouse T cells, he found that DNA methylation increased in large regions of the genome as expected. However, these same cancer samples possessed equally large genomic regions which had lost DNA methylation when compared with normal, non-cancerous T cells.
“This was surprising because the dogma for the last ten years has been that TET mutations lead to increased DNA methylation,” said López-Moyado. “While this is correct for certain genomic regions, it is not true for large swaths of the genome known as heterochromatin.” Heterochromatin is a form of DNA that is tightly packed, usually inactive and typically resides in the periphery of the nucleus.
A closer look at the distribution of Dnmt3a molecules in TET-deficient cells showed that a portion of Dnmt3a molecules had relocated away from heterochromatin, explaining the seemingly paradoxical loss of methylation in this region of the genome.
Interestingly, a substantial number of leukemia patients bear both TET2 and DNMT3A mutations and mice that carry dual mutations in both TET2 and DNMT3A are known to develop more severe cancers than mice with TET2 or DNMT3A mutations alone. This observation prompted López-Moyado to analyze published data from mouse hematopoietic stem cells—the cells from which blood cancers arise—to establish whether double mutations correlate with changes in the methylation status of heterochromatin.
López-Moyado found that cells with dual TET2 and DNMT3A mutations showed an even more pronounced loss of DNA methylation in heterochromatin than cells with only one of these mutations. In fact, even without mutations in TET or DNMT3A, decreased DNA methylation in heterochromatin is common in cancer, suggesting that loss of TET or DNMT function may be involved.
Heterochromatin constitutes more than half of mammalian genomes, and contains not only the genes that need to be silenced in any particular cell type, but also “parasitic” DNA elements and various “repeat” sequence that long ago invaded the genome. These DNA elements, which include transposons and ancient viruses, need to be stringently repressed in heterochromatin.
“If heterochromatin loses DNA methylation, these elements can become reactivated and can form aberrant structures or jump from one genomic location to another, leading to genome instability and DNA damage, the hallmarks of cancer,” explains López-Moyado. Indeed, these features are commonly observed, together with DNA hypomethylation, in many types of hereditary as well as non-hereditary cancers, including cancers with TET and DNMT3A mutations.
Future research will establish if the loss of DNA methylation in heterochromatin that results from mutations in the DNA methylation pathway has a direct role in heterochromatin reactivation and cancer initiation and progression.
Full citation: Isaac F. López-Moyado, Ageliki Tsagaratou, Hiroshi Yuita, Hyungseok Seo, Benjamin Delatte, Sven Heinz, Christopher Benner, Anjana Rao. Proc Natl Acad Sci USA 2019;
The study was funded by NIH grant R35 CA210043 (to A.R.); a graduate student fellowship from CONACYT/ UCMEXUS (to I.F. L.-M.); equipment grants S10OD016262 and S10 RR027366 to LJI; the Salk Institute’s NGS Core Facility with funding from NIH-NCI CCSG: P30 014195, the Chapman Foundation and the Helmsley Charitable Trust; Irvington Institute postdoctoral fellowships from the Cancer Research Institute (to A.T. and H.S.); an AACR-Genentech Immuno-oncology Research Fellowship (to H.S.); and postdoctoral fellowship from the Jane Coffin Childs Memorial Fund (to B.D.).


